Introduction to the integration hands-on
The Hitchhiker’s Guide to scRNA-seq, iMM
António Sousa
09/07/2024
Seurat
R lists
A list in R is an object that can comprise different data types of different lengths. The elements in the list are ordered and changeable.
# set seed
set.seed(123)
# student class sizes
student_class_sizes <- sample(15:30, 3)
# list with 3 classes with student class names
student_class_names <- list("class1" = randomNames::randomNames(n = student_class_sizes[1]),
"class2" = randomNames::randomNames(n = student_class_sizes[2]),
"class3" = randomNames::randomNames(n = student_class_sizes[3]))Lists can be queried or indexed by name or index position number in the list.
## [1] "Alexander, Olivia" "Garcia, Norman" "al-Badour, Naafoora"
## [4] "Rosen, Stephanie" "Tilmon, Nicolette" "Ledbetter, Matthew"## [1] "Chong, David" "Soukup, Austin" "Jeffers, Megan"
## [4] "Culhane, Tiffany" "Cao, Lars" "Worley, Christopher"lapply function
lapply is a function that applies a function to every element in a list returning the result into a list.
# check the size/length of every class in the list
class_size <- lapply(student_class_names, length)
class_size## $class1
## [1] 29
##
## $class2
## [1] 30
##
## $class3
## [1] 17The FUN parameter can take any existing function or a customized function.
# get the length, i.e., no. of characters, of the longest name in every class
longest_name <- lapply(X = student_class_names, FUN = function(x) {
max( nchar(x) ) # nested functions
})
longest_name## $class1
## [1] 21
##
## $class2
## [1] 20
##
## $class3
## [1] 19dplyr syntax
##
## Attaching package: 'dplyr'## The following objects are masked from 'package:stats':
##
## filter, lag## The following objects are masked from 'package:base':
##
## intersect, setdiff, setequal, union## Sepal.Length Sepal.Width Petal.Length Petal.Width Species
## 1 5.1 3.5 1.4 0.2 setosa
## 2 4.9 3.0 1.4 0.2 setosa
## 3 4.7 3.2 1.3 0.2 setosa
## 4 4.6 3.1 1.5 0.2 setosa
## 5 5.0 3.6 1.4 0.2 setosa
## 6 5.4 3.9 1.7 0.4 setosa## Sepal.Length Sepal.Width Petal.Length Petal.Width
## 1 5.843333 3.057333 3.758 1.199333# get the mean of 'Sepal.Length' when 'Petal.Length' is higher than 3.7
iris %>%
filter(Petal.Length > 3.7) %>%
summarise(Sepal.Length.Mean = mean(Sepal.Length))## Sepal.Length.Mean
## 1 6.337634Input
10X scRNA-seq data processed with CellRanger software can be imported into R/Seurat by specifying the folder holding the following three files (they can be compressed as well): barcodes.tsv, matrix.tsv, genes.tsv.
# 10X data directory (downloaded from 10X website - see links below):
#https://support.10xgenomics.com/single-cell-gene-expression/datasets/1.1.0/pbmc6k
#https://support.10xgenomics.com/single-cell-gene-expression/datasets/2.1.0/pbmc8k
data10X.dir <- "../hands-on/data/10x_3pV1_2"
samples.10X <- c("pbmc6k_3pV1", "pbmc8k_3pV2")
samples.10X.dir <- file.path(data10X.dir, samples.10X)
names(samples.10X.dir) <- c("V1", "V2")
samples.10X.dir## V1
## "../hands-on/data/10x_3pV1_2/pbmc6k_3pV1"
## V2
## "../hands-on/data/10x_3pV1_2/pbmc8k_3pV2"## [1] "barcodes.tsv" "barcodes.tsv" "genes.tsv" "genes.tsv" "matrix.mtx"
## [6] "matrix.mtx"Import 10X samples from the same project into Seurat.
Import 10X samples from the different projects, i.e., processed independently with CellRanger, into Seurat.
# import 10X samples as individual 10X samples
seu.list <- lapply(X = samples.10X.dir, FUN = function(x) {
CreateSeuratObject(counts = Read10X(data.dir = x))
})## Warning: Feature names cannot have underscores ('_'), replacing with dashes
## ('-')
## Warning: Feature names cannot have underscores ('_'), replacing with dashes
## ('-')# merge samples & counts layer
seu <- merge(x = seu.list[[1]], y = seu.list[[2]], add.cell.ids = names(seu.list))
seu[["RNA"]] <- JoinLayers(object = seu[["RNA"]])
seu## An object of class Seurat
## 36116 features across 13800 samples within 1 assay
## Active assay: RNA (36116 features, 0 variable features)
## 1 layer present: countsExport a Seurat object in RDS format.
# export Seurat R object as .RDS file
saveRDS(object = seu, file = file.path(data10X.dir, "seurat.rds"))Import a Seurat object saved as RDS format.
SeuratObject
SeuratObject
SeuratObject is a S4 class object. The different layers of information can be accessed with Seurat functions or using the accessor @ (see examples below).
@assays
## $RNA
## Assay (v5) data with 36116 features for 13800 cells
## First 10 features:
## MIR1302-10, FAM138A, OR4F5, RP11-34P13.7, RP11-34P13.8, AL627309.1,
## RP11-34P13.14, RP11-34P13.9, AP006222.2, RP4-669L17.10
## Layers:
## counts@meta.data
## orig.ident nCount_RNA nFeature_RNA
## V1_AAACATACAACCAC-1 SeuratProject 2029 642
## V1_AAACATACACCAGT-1 SeuratProject 2835 1031
## V1_AAACATACCCGTAA-1 SeuratProject 1804 688
## V1_AAACATACCTAAGC-1 SeuratProject 1789 621
## V1_AAACATACCTTCCG-1 SeuratProject 3155 928
## V1_AAACATACGCGAAG-1 SeuratProject 3416 1190Cell metadata can be added to the seu@meta.data slot.
# Add more meta data:
#import "Source Data Fig.3" excel file with cell annotations from Harmony paper: https://doi.org/10.1038/s41592-019-0619-0
meta.data <- readxl::read_excel(path = file.path(data10X.dir, "41592_2019_619_MOESM9_ESM.xlsx"))
meta.data <- dplyr::filter(meta.data, dataset %in% c("three_prime_v1", "three_prime_v2"))
meta.data <- as.data.frame(meta.data)
row.names(meta.data) <- paste(paste0(toupper(gsub("three_prime_", "", meta.data$dataset)), "_"),
meta.data$cell_id, "-1", sep = "")
meta.data <- meta.data[! is.na(meta.data$cell_subtype), ] # remove cells not annotated
seu <- subset(seu, cells = row.names(meta.data)) # removed 754 cells (total: 13,046)
seu$cell_type <- meta.data[colnames(seu), "cell_subtype"] # adding cell annotations to Seurat meta data
seu$batch <- ifelse(grepl(pattern = "^V1_", x = colnames(seu)), "V1", "V2")## orig.ident nCount_RNA nFeature_RNA cell_type batch
## V1_AAACATACAACCAC-1 SeuratProject 2029 642 cd8naive V1
## V1_AAACATACACCAGT-1 SeuratProject 2835 1031 mono14 V1
## V1_AAACATACCCGTAA-1 SeuratProject 1804 688 cd8naive V1
## V1_AAACATACCTAAGC-1 SeuratProject 1789 621 cd4naive V1
## V1_AAACATACCTTCCG-1 SeuratProject 3155 928 mono14 V1
## V1_AAACATACGCGAAG-1 SeuratProject 3416 1190 bmem V1@graphs/neighbors/reductions
Standard workflow
Standard workflow
Dimensional reduction
## Plot jointly dimreds
# Run UMAP
seu <- RunUMAP(seu, dims = 1:30, reduction = "pca", reduction.name = "umap.unintegrated")
# Plot
unint.dimreds <- list()
unint.dimreds[["pca_batch"]] <- DimPlot(seu, reduction = "pca", group.by = "batch", pt.size = 0.1, alpha = 0.5)
unint.dimreds[["pca_cell_type"]] <- DimPlot(seu, reduction = "pca", group.by = "cell_type", pt.size = 0.1, alpha = 0.5)
unint.dimreds[["umap_batch"]] <- DimPlot(seu, reduction = "umap.unintegrated", group.by = "batch", pt.size = 0.1, alpha = 0.5)
unint.dimreds[["umap_cell_type"]] <- DimPlot(seu, reduction = "umap.unintegrated", group.by = "cell_type", pt.size = 0.1, alpha = 0.5)Batch assessment (PCA)
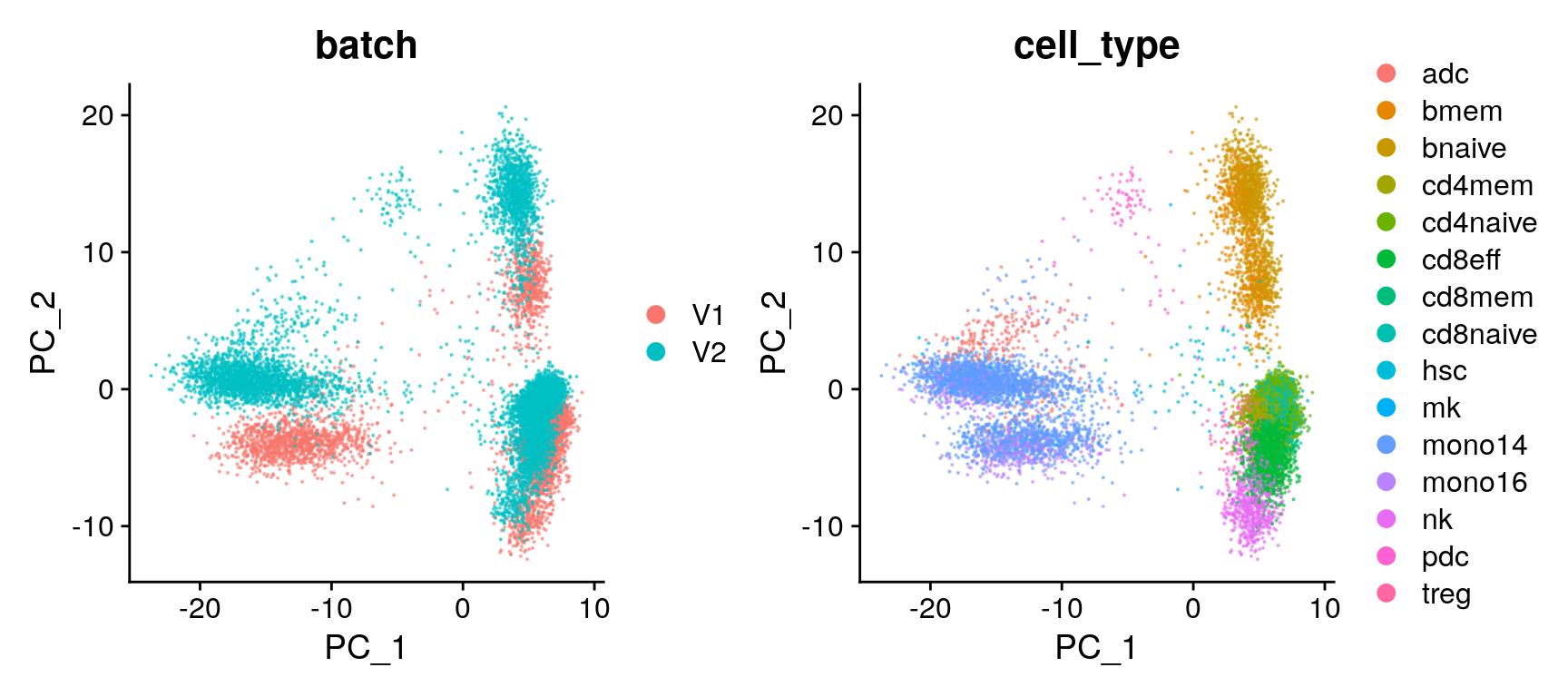
Batch assessment (UMAP)
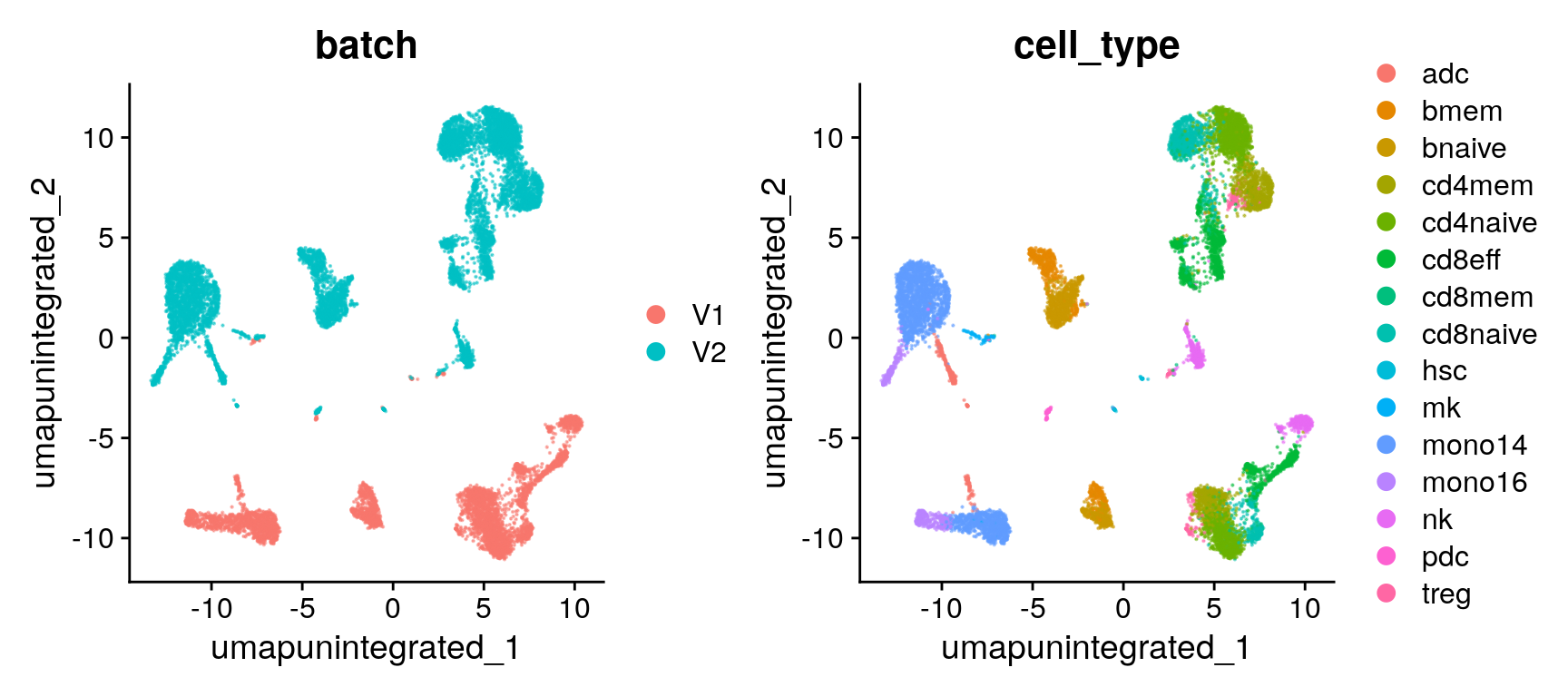
Clustering
# graph-based clustering
seu <- FindNeighbors(seu, dims = 1:30, reduction = "pca") # shared nearest-neighbor graph
seu <- FindClusters(seu, resolution = 2, cluster.name = "unintegrated_clusters") # cell population detection - Louvain algorithm## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 13046
## Number of edges: 558289
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.8521
## Number of communities: 32
## Elapsed time: 1 seconds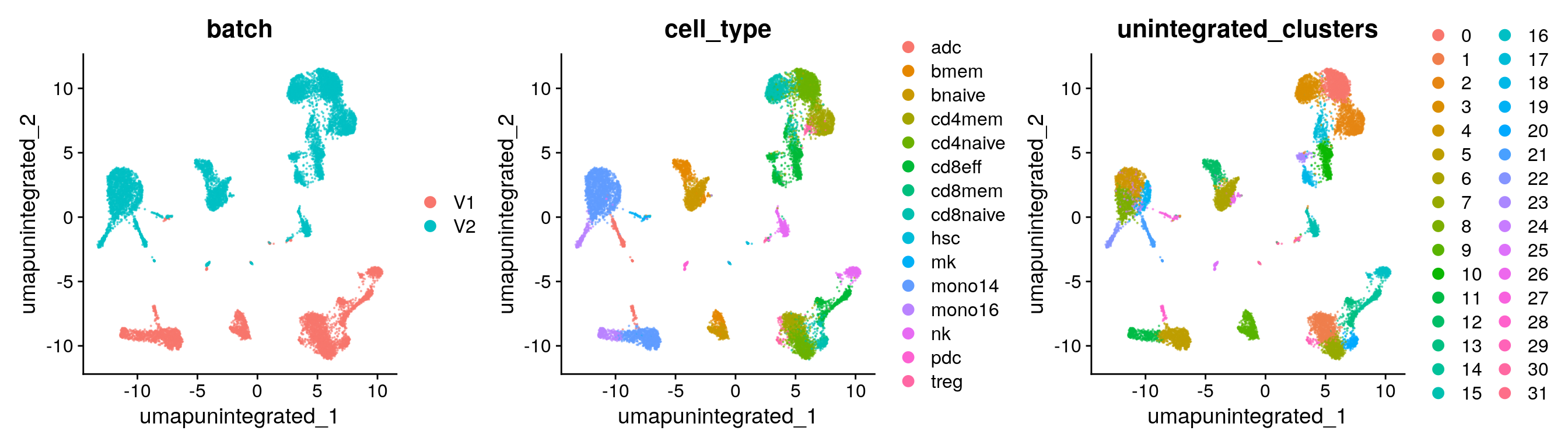
Integration
Split layers
Split layers before integration by providing the cell batch label identity.
## An object of class Seurat
## 36116 features across 13046 samples within 1 assay
## Active assay: RNA (36116 features, 2000 variable features)
## 3 layers present: counts, data, scale.data
## 2 dimensional reductions calculated: pca, umap.unintegrated## Splitting 'counts', 'data' layers. Not splitting 'scale.data'. If you would like to split other layers, set in `layers` argument.## An object of class Seurat
## 36116 features across 13046 samples within 1 assay
## Active assay: RNA (36116 features, 2000 variable features)
## 5 layers present: counts.V1, counts.V2, scale.data, data.V1, data.V2
## 2 dimensional reductions calculated: pca, umap.unintegratedIntegration workflow
Different integration methods can be called from Seurat IntegrateLayers() function: CCAIntegration and RPCAIntegration (from Seurat), HarmonyIntegration, FastMNNIntegration and scVIIntegration (wrapper functions from SeuratWrappers - the packages from the respective methods need to be installed first).
# integration Seurat workflow
seu <- NormalizeData(seu)
seu <- FindVariableFeatures(seu)
seu <- ScaleData(seu)
seu <- RunPCA(seu)
seu <- IntegrateLayers(object = seu,
method = CCAIntegration, # RPCAIntegration / HarmonyIntegration / FastMNNIntegration / scVIIntegration
orig.reduction = "pca",
new.reduction = "integrated.cca")
seu[["RNA"]] <- JoinLayers(seu[["RNA"]])Clustering
# cluster integrated dimensional reduction
seu <- FindNeighbors(seu, reduction = "integrated.cca", dims = 1:30)
seu <- FindClusters(seu, resolution = 2, cluster.name = "integrated_clusters")## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 13046
## Number of edges: 641240
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.7902
## Number of communities: 28
## Elapsed time: 2 seconds## An object of class Seurat
## 36116 features across 13046 samples within 1 assay
## Active assay: RNA (36116 features, 2000 variable features)
## 3 layers present: data, counts, scale.data
## 3 dimensional reductions calculated: pca, umap.unintegrated, integrated.ccaDimensional reduction
## Plot jointly dimreds
# Run UMAP
seu <- RunUMAP(seu, dims = 1:30, reduction = "integrated.cca", reduction.name = "umap.integrated")
# Plot
int.dimreds <- list()
int.dimreds[["umap_batch"]] <- DimPlot(seu, reduction = "umap.integrated", group.by = "batch", pt.size = 0.1, alpha = 0.5)
int.dimreds[["umap_cell_type"]] <- DimPlot(seu, reduction = "umap.integrated", group.by = "cell_type", pt.size = 0.1, alpha = 0.5)
int.dimreds[["umap_clusters"]] <- DimPlot(seu, reduction = "umap.integrated", group.by = "integrated_clusters", pt.size = 0.1, alpha = 0.5)
int.dimreds[[1]] + int.dimreds[[2]] + int.dimreds[[3]]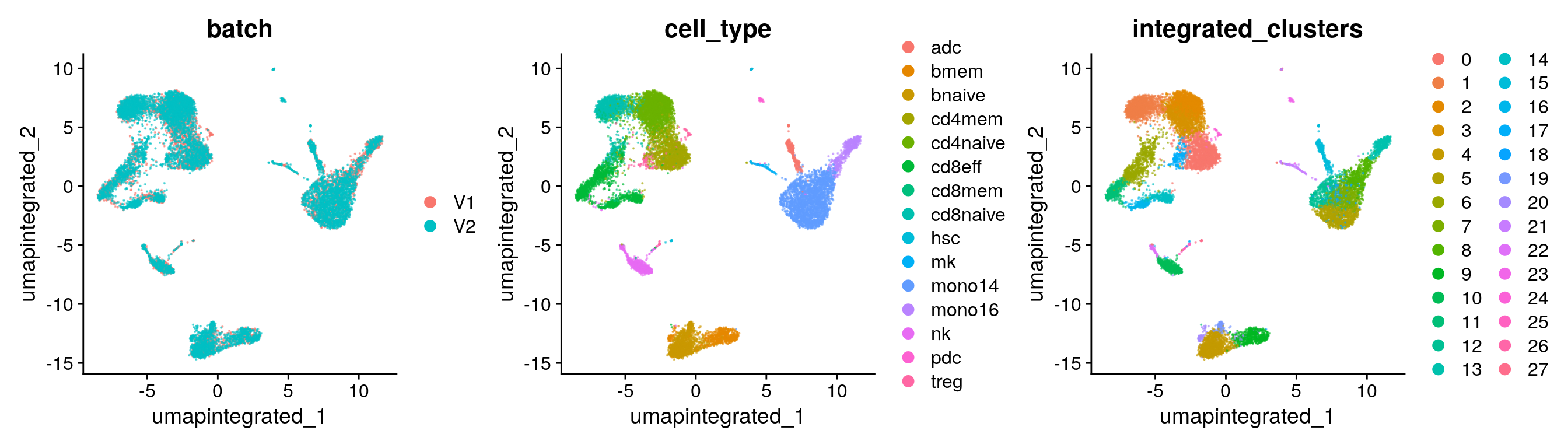
Reference-mapping
Azimuth references
SeuratData provides some ready to use Azimuth references.
## Reference-mapping with Azimuth
# Import library
library("Azimuth")
library("SeuratData") # contains some Azimuth references
# Azimuth references available in SeuratData
SeuratData::AvailableData() %>% filter(grepl("ref.", row.names(.), ))## Dataset Version Summary
## adiposeref.SeuratData adiposeref 1.0.0 Azimuth Reference: adipose
## bonemarrowref.SeuratData bonemarrowref 1.0.0 Azimuth Reference: bonemarrow
## fetusref.SeuratData fetusref 1.0.0 Azimuth Reference: fetus
## heartref.SeuratData heartref 1.0.0 Azimuth Reference: heart
## humancortexref.SeuratData humancortexref 1.0.0 Azimuth Reference: humancortex
## kidneyref.SeuratData kidneyref 1.0.2 Azimuth Reference: kidney
## lungref.SeuratData lungref 2.0.0 Azimuth Reference: lung
## mousecortexref.SeuratData mousecortexref 1.0.0 Azimuth Reference: mousecortex
## pancreasref.SeuratData pancreasref 1.0.0 Azimuth Reference: pancreas
## pbmcref.SeuratData pbmcref 1.0.0 Azimuth Reference: pbmc
## tonsilref.SeuratData tonsilref 2.0.0 Azimuth Reference: tonsil
## species system ncells tech
## adiposeref.SeuratData human adipose 160075 scRNA-seq and sNuc-seq
## bonemarrowref.SeuratData human bonemarrow 297627 10x v2
## fetusref.SeuratData human fetus 377456 <NA>
## heartref.SeuratData human heart 656509 scRNA-seq and sNuc-seq
## humancortexref.SeuratData human motor cortex 76533 <NA>
## kidneyref.SeuratData human kidney 64693 snRNA-seq
## lungref.SeuratData human lung 584884 <NA>
## mousecortexref.SeuratData mouse motor cortex 159738 10x v3
## pancreasref.SeuratData human pancreas 35289 <NA>
## pbmcref.SeuratData human PBMC 2700 10x v1
## tonsilref.SeuratData human tonsil 377963 scRNA-seq
## seurat default.dataset disk.datasets other.datasets
## adiposeref.SeuratData <NA> <NA> <NA> <NA>
## bonemarrowref.SeuratData <NA> <NA> <NA> <NA>
## fetusref.SeuratData <NA> <NA> <NA> <NA>
## heartref.SeuratData <NA> <NA> <NA> <NA>
## humancortexref.SeuratData <NA> <NA> <NA> <NA>
## kidneyref.SeuratData <NA> <NA> <NA> <NA>
## lungref.SeuratData <NA> <NA> <NA> <NA>
## mousecortexref.SeuratData <NA> <NA> <NA> <NA>
## pancreasref.SeuratData <NA> <NA> <NA> <NA>
## pbmcref.SeuratData <NA> <NA> <NA> <NA>
## tonsilref.SeuratData <NA> <NA> <NA> <NA>
## notes Installed InstalledVersion
## adiposeref.SeuratData <NA> FALSE <NA>
## bonemarrowref.SeuratData <NA> FALSE <NA>
## fetusref.SeuratData <NA> FALSE <NA>
## heartref.SeuratData <NA> FALSE <NA>
## humancortexref.SeuratData <NA> FALSE <NA>
## kidneyref.SeuratData <NA> FALSE <NA>
## lungref.SeuratData <NA> FALSE <NA>
## mousecortexref.SeuratData <NA> FALSE <NA>
## pancreasref.SeuratData <NA> FALSE <NA>
## pbmcref.SeuratData <NA> TRUE 1.0.0
## tonsilref.SeuratData <NA> FALSE <NA>Azimuth reference-mapping
Azimuth can be run by providing the Seurat object to be annotated and one of the SeuratData Azimuth references.
## Azimuth reference-mapping
refmap <- RunAzimuth(seu, reference = "pbmcref") # installs 'pbmcref' if does not exists
refmap## An object of class Seurat
## 36211 features across 13046 samples within 4 assays
## Active assay: RNA (36116 features, 2000 variable features)
## 3 layers present: data, counts, scale.data
## 3 other assays present: prediction.score.celltype.l1, prediction.score.celltype.l2, prediction.score.celltype.l3
## 6 dimensional reductions calculated: pca, umap.unintegrated, integrated.cca, umap.integrated, integrated_dr, ref.umapProject query onto reference
## project reference onto query
refmap.dimreds <- list()
refmap.dimreds[["umap_batch"]] <- DimPlot(refmap, reduction = "ref.umap", group.by = "batch", label = TRUE, pt.size = 0.1, alpha = 0.5) + NoLegend()
refmap.dimreds[["umap_cell_type"]] <- DimPlot(refmap, reduction = "ref.umap", group.by = "cell_type", label = TRUE, pt.size = 0.1, alpha = 0.5) + NoLegend()
refmap.dimreds[["umap_preds_l1"]] <- DimPlot(refmap, reduction = "ref.umap", group.by = "predicted.celltype.l1", label = TRUE, pt.size = 0.1, alpha = 0.5) + NoLegend()
refmap.dimreds[["umap_preds_l2"]] <- DimPlot(refmap, reduction = "ref.umap", group.by = "predicted.celltype.l2", label = TRUE, pt.size = 0.1, alpha = 0.5) + NoLegend()
refmap.dimreds[["umap_preds_l3"]] <- DimPlot(refmap, reduction = "ref.umap", group.by = "predicted.celltype.l3", label = TRUE, pt.size = 0.1, alpha = 0.5) + NoLegend()
refmap.dimreds[[1]] + refmap.dimreds[[2]] + refmap.dimreds[[3]] + refmap.dimreds[[4]] + refmap.dimreds[[5]] + patchwork::plot_layout(ncol = 5)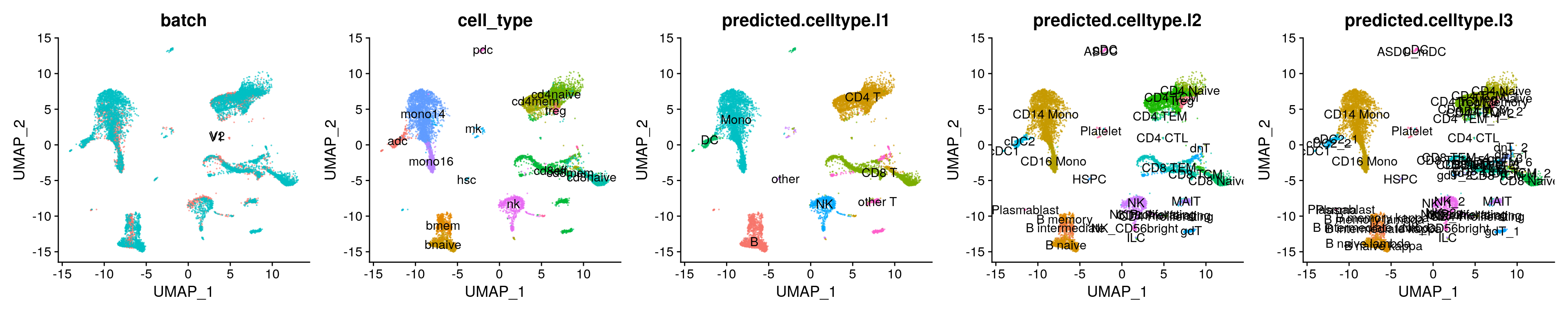
Custom reference-mapping
Custom reference-mapping can be performed by calling the Seurat functions FindTransferAnchors() and MapQuery().
## customized reference
# split Seurat object into reference and query
seu.list <- SplitObject(object = seu, split.by = "batch")
# Normalize reference and query
seu.list <- lapply(seu.list, NormalizeData)
# Process the reference
seu.list$V2 <- FindVariableFeatures(seu.list$V2)
seu.list$V2 <- ScaleData(seu.list$V2)
seu.list$V2 <- RunPCA(seu.list$V2)
seu.list$V2 <- RunUMAP(seu.list$V2, dims = 1:30, reduction = "pca", return.model = TRUE)
# Transfer anchors between reference and query
anchors <- FindTransferAnchors(reference = seu.list$V2, query = seu.list$V1,
dims = 1:30, reference.reduction = "pca")
# Map query
seu.list$V1 <- MapQuery(anchorset = anchors, reference = seu.list$V2, query = seu.list$V1,
refdata = list(celltype = "cell_type"), reference.reduction = "pca",
reduction.model = "umap")Project query onto custom reference
## project reference onto query
custom.refmap.dimreds <- list()
custom.refmap.dimreds[["ref_umap_cell_type"]] <- DimPlot(seu.list$V2, reduction = "umap", group.by = "cell_type",
label = TRUE, pt.size = 0.1, alpha = 0.5) + NoLegend()
custom.refmap.dimreds[["query_umap_cell_type"]] <- DimPlot(seu.list$V1, reduction = "ref.umap", group.by = "cell_type", label = TRUE, pt.size = 0.1, alpha = 0.5) + NoLegend()
custom.refmap.dimreds[["query_umap_preds"]] <- DimPlot(seu.list$V1, reduction = "ref.umap", group.by = "predicted.celltype", label = TRUE, pt.size = 0.1, alpha = 0.5) + NoLegend()
custom.refmap.dimreds[[1]] + custom.refmap.dimreds[[2]] + custom.refmap.dimreds[[3]]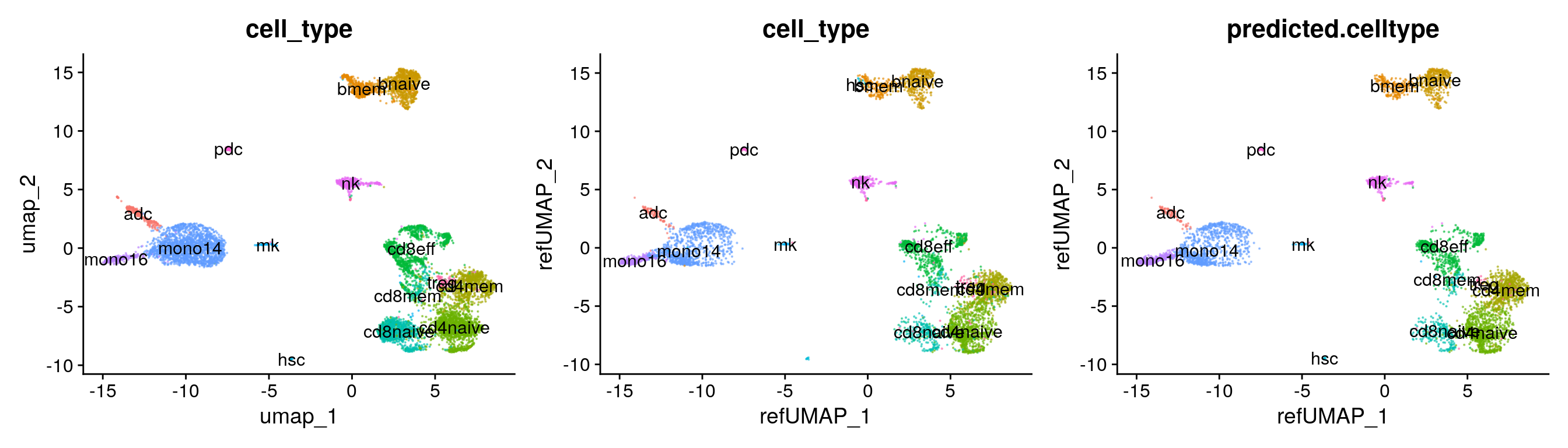
Custom reference-mapping assessment
Confusion matrix between predicted cell type labels versus ground-truth.
##
## adc bmem bnaive cd4mem cd4naive cd8eff cd8mem cd8naive hsc mk
## adc 72 0 0 0 0 0 0 0 0 0
## bmem 0 183 25 0 0 0 0 0 12 0
## bnaive 0 11 361 0 0 0 0 0 0 0
## cd4mem 0 0 0 567 127 6 12 13 0 0
## cd4naive 0 0 0 26 851 0 3 35 0 0
## cd8eff 0 1 0 4 1 523 15 11 0 0
## cd8mem 0 0 0 0 0 1 0 0 0 0
## cd8naive 0 0 0 1 4 20 24 240 0 0
## hsc 0 0 0 0 0 0 0 0 8 0
## mk 0 0 0 0 0 0 0 0 0 19
## mono14 3 1 0 0 0 0 0 0 0 3
## mono16 1 0 0 0 0 0 0 0 0 0
## nk 0 0 0 0 0 2 1 0 0 0
## pdc 0 0 0 0 0 0 0 0 0 0
## treg 0 1 0 2 0 1 0 0 1 0
##
## mono14 mono16 nk pdc treg
## adc 4 0 0 0 0
## bmem 0 0 0 0 0
## bnaive 0 0 0 0 0
## cd4mem 0 0 0 0 26
## cd4naive 0 0 0 0 6
## cd8eff 0 0 13 0 1
## cd8mem 0 0 0 0 0
## cd8naive 0 0 0 0 0
## hsc 0 0 0 0 0
## mk 0 0 0 0 0
## mono14 803 6 1 0 0
## mono16 50 326 0 0 0
## nk 0 0 281 0 0
## pdc 0 0 0 11 0
## treg 0 0 1 0 49